
Il
termine EPILESSIA definisce un disturbo neurologico cronico caratterizzato dal
verificarsi periodico e imprevedibile di CRISI EPILETTICHE, cioè alterazioni
transitorie del comportamento conseguenti a una scarica patologica,
ipersincrona e ritmica di una popolazione estesa di neuroni cerebrali di tipo
eccitatorio che riescono a superare la barriera data invece dai neuroni
inibitori.
Poiché
l’epilessia può generarsi in qualsiasi punto del SNC nei cosiddetti focus epilettici, cioè punti di partenza
della scarica elettrica patologica, di varia origine e non sempre individuabili,
le manifestazioni dipenderanno dall’area corticale da cui si origina la
scarica, dalle funzioni di questa area e dall’eventuale diffusione (spreading) che avviene quando la scarica
elettrica del focus oltrepassa la soglia attivando anche i neuroni circostanti:
ad esempio, se il focus coinvolge la corteccia cerebrale fondamentale per la
memoria immediata, i movimenti, il linguaggio, la vista, l’udito, l’attivazione
darà una convulsione; se, invece, il bersaglio è l’ipotalamo, le ripercussioni
si avranno sul sistema autonomo periferico con alterazioni a livello simpatico
e parasimpatico, mentre le scariche sul midollo allungato produrranno una
perdita di coscienza.
Il
5% di tutte le persone ha almeno una crisi epilettica durante la sua vita, ma
non è considerato affetto da epilessia, in quanto la diagnosi implica una
tendenza a crisi epilettiche ripetute che si trova nello 0.5% della
popolazione.
Per
la diagnosi di epilessia è necessaria un'accurata valutazione dei sintomi e
della storia clinica, che deve possibilmente comprendere anche le osservazioni
dettagliate da parte di terzi, in quanto l'alterazione o la perdita di
coscienza spesso precludono una descrizione dei sintomi da parte del paziente
stesso. L'elettroencefalogramma (EEG) rileva l'attività elettrica del cervello
ed è un'analisi fondamentale nella diagnosi dell'epilessia, perché le
alterazioni elettriche, spesso molto indicative, possono essere presenti anche
in assenza dei sintomi. Al di fuori delle crisi epilettiche, però, le
alterazioni elettriche possono mancare, pertanto un EEG normale registrato al
di fuori di una crisi non esclude la diagnosi di epilessia. Altri esami
diagnostici includono la risonanza magnetica o TAC cerebrale ed esami di
laboratorio, e sono indicati per accertare o escludere cause specifiche.
La
classificazione di questa malattia, può essere fatta in base all’eziologia: si parla di epilessia primaria o idiopatica
quando la storia clinica e gli esami diagnostici non rivelano cause per crisi
epilettiche ripetute e di epilessia secondaria o sintomatica quando, invece,
può essere identificata la sua eziologia (lesioni cerebrali, neoplasie,
infezioni, intossicazioni); mentre la maggior parte delle epilessie idiopatiche
è infatti dovuta a fattori genetici e metabolici ancora sconosciuti e si
manifesta in età infantile o adolescente, una grande parte delle epilessie
secondarie si manifesta dopo i 40 anni.
Tuttavia
la classificazione clinica è quella maggiormente impiegata e prevede due
principali categorie: le crisi (o accessi) parziali e le crisi (o accessi)
generalizzate, ciascuna delle quali distinguibili ulteriormente in semplice
quando non si ha perdita di conoscenza e complessa quando la perdita di
coscienza è immediata.
Le
CRISI PARZIALI rappresentano il 60% di tutte le epilessie, hanno origine focale
a livello della corteccia, cioè la scarica inizia e generalmente rimane
circoscritta in una regione cerebrale di un solo emisfero, e determinano
soltanto contrazioni muscolari involontarie con esperienze sensoriali anomale
della durata di pochi secondi, senza perdita di coscienza. Quando si ha perdita
di coscienza si parla di CRISI PARZIALI COMPLESSE che frequentemente
interessano i due emisferi: in genere
hanno origine nel lobo temporale e sono caratterizzate da una perdita di
coscienza variabile tra 30 sec e 2 minuti, associata spesso a movimenti
afinalistici come lo schioccare delle labbra e/o la torsione della mano.
Le
crisi parziali sono possibili a tutte le età, ma sono più frequenti nella vecchiaia,
inoltre, tutti gli accessi parziali possono trapassare in accessi
generalizzati, cosicché la loro diagnosi riesce difficile.
Al
contrario delle crisi parziali che originano in aree localizzate della
corteccia, nelle CRISI GENERALIZZATE è impossibile precisare l’area anatomica.
In genere, originano dalla scarica reciproca del talamo e della corteccia e gli
attacchi sono accompagnati già all’inizio dalle riduzione della coscienza.
Si
distinguono in:
- ASSENZE o PICCOLO MALE caratterizzata da scariche elettriche di breve durata che determinano fugati perdite di coscienza (20 sec) senza dare convulsioni: in pratica il soggetto sembra assente, ha lo sguardo fisso, rimane immobile e non risponde agli stimoli. Dopo questa breve interruzione, il soggetto riprende l'attività senza avvertire la breve sospensione dello stato di coscienza.
- GRANDE MALE o CRISI TONICO-CLONICA è caratterizzata da scariche abnormi di tutto il SNC che determinano una iniziale e intensa contrazione di tutta la muscolatura con coinvolgimento anche del sistema nervoso autonomo che si manifesta con scialorrea (bava) e perdita del controllo degli sfinteri (minzione e defecazione): tale fase "tonica" che può essere anche dolorosa (anche se non avvertita dal paziente vista la perdita di coscienza) è seguita da una fase "clonica" in cui si hanno contrazioni alternate a rilasciamento. L'esito di questa forma di epilessia può essere addirittura infausto per soffocamento (chiusura dell'epiglottide dovuta alla retrazione dell'ipoglosso) o per fibrillazione (scarica abnorme sui recettori cardiaci adrenergici β1)
Le
anomalie neuronali responsabili dell’epilessia non sono state ancora
perfettamente chiarite ma, alla luce del ruolo fondamentale che hanno le
sinapsi nel mediare la comunicazione tra i neuroni cerebrali, si è ipotizzato
che una disfunzione sinaptica possa essere responsabile di una crisi epilettica.
Il
TRATTAMENTO FARMACOLOGICO dell’epilessia è esclusivamente sintomatico, potendo
esso controllare la comparsa delle crisi, mentre non è disponibile una terapia
curativa. Tuttavia
garantisce una vita normale a molti pazienti che altrimenti sarebbero
gravemente limitati o minacciati da frequenti crisi epilettiche.
In
linea teorica un farmaco antiepilettico ideale dovrebbe sopprimere le crisi
senza causare effetti indesiderati ma sfortunatamente i farmaci attualmente
usati non solo non controllano le crisi in tutti i pazienti ma causano
frequentemente effetti indesiderati per cui il problema maggiore nella terapia
antiepilettica è rappresentato dalla costanza nell’assunzione dei farmaci vista
la necessità di terapie a lungo termine.
La
terapia deve tenere conto della situazione e delle esigenze individuali del
paziente e va indicata con cura, perché è prolungata e con effetti collaterali
potenzialmente gravi, che possono comunque essere minimizzati nella maggior
parte dei casi. In linea generale, dopo aver effettuato la diagnosi di una
crisi epilettica in una persona, il medico dovrebbe fare un tentativo nel
cercare di determinare la causa nella speranza di individuare una lesione
strutturale e/o metabolica curabile. Fallito tale tentativo, il medico dovrebbe
chiedersi se e quando iniziare la terapia: ad esempio, la terapia
antiepilettica potrebbe esser non necessaria nel caso in cui si abbia avuto un
attacco tonico-clonico isolato in un adulto giovane e sano senza casi di
epilessia in famiglia e con esami neurologici normali, dato che la probabilità
che la crisi epilettica si ripeta negli anni successivi (15%) è
approssimativamente simile al rischio di sviluppare una reazione ai farmaci che
induca l’interruzione della cura. Al contrario, il verificarsi di un simile
attacco convulsivo in un individuo con una storia famigliare di epilessia,
esami neurologici anormali, comporta un rischio di ricorrenza del 60% che depone
a favore dell’inizio della terapia.
Pertanto,
il medico avrà come obiettivo primario la scelta del farmaco più efficace nel
controllo della crisi, cercando di mantenere gli effetti collaterali a un
livello accettabile. Per minimizzare la tossicità, la cura andrebbe iniziata
con un solo farmaco ad un dosaggio ridotto e aumentando la dose basandosi sul
controllo della crisi e sulla comparsa di effetti tossici. Se, invece, si
verifica una crisi nonostante i livelli di farmaco siano ottimali, andrebbe prima
valutata l’eventuale presenza di fattori potenzialmente scatenanti
(deprivazione di sonno, febbre, farmaci contenenti sostanze che abbassano la
soglia delle crisi epilettiche come la caffeina) e poi eventualmente cambiare
farmaco, diminuendo gradualmente il dosaggio del primo in modo da ridurre il
rischio di crisi. Nell’eventualità che anche questo secondo farmaco risulti
inadeguato, si può ricorrere alla somministrazione contemporanea di due o più
farmaci che agiscano tramite meccanismi diversi.
La
durata della terapia antiepilettica dipende dal tipo, dalla causa e dall’evoluzione
spontanea dell’epilessia. Generalmente si propone una graduale riduzione dei
farmaci quando per 2-5 anni non si sono verificate crisi epilettiche e quando
sono assenti o minime le alterazioni dell’EEG. La sospensione deve avvenire
gradualmente per evitare fenomeni di rebound e di status epilepticus che si
hanno con la sospensione brusca e in genere nell’80% dei casi le crisi
riappaiono entro 6 mesi dopo la sospensione con la conseguente necessità di
riprendere la terapia. La prognosi è migliore quando le crisi sono infrequenti
e controllate con basse dosi di un farmaco.
Inoltre,
poiché l’epilessia interessa in molti casi l’età riproduttiva, l’impiego di
farmaci antiepilettici ha diverse complicazioni per la salute della donna.
Innanzitutto,
la maggior parte dei farmaci antiepilettici, essendo induttori del CYP450,
diminuiscono l’efficacia dei contraccettivi orali, per aumento del loro
metabolismo epatico.
A
ciò si aggiungono gli effetti teratogeni e il fatto che gli stessi farmaci che
inducono il CYP sono stati associati all’induzione di un deficit di vitamina K
nel neonato con conseguente coagulopatia e emorragia intracerebrale che determina
la raccomandazione di assumere come profilassi 10 mg di vitamina K durante
l’ultimo mese di gestazione. Pertanto, una possibilità per una donna epilettica
che desideri avere una gravidanza, è tentare un periodo senza farmaci o in
alternativa usare solo un farmaco tenendo sotto controllo la concentrazione
plasmatica.
I
meccanismi d’azione dei farmaci antiepilettici sono principalmente tre:
potenziamento dell’azione del GABA, inibizione della funzionalità dei canali
del Na+ e inibizione della funzionalità dei canali del Ca+2.
I
farmaci efficaci nella terapia delle forme più comuni di epilessia, cioè le
forme parziali e tonico-cloniche sembrano agire attraverso i primi due
meccanismi, mentre i farmaci efficaci nella terapia delle assenze utilizzano il
terzo.
La
comprensione dei meccanismi delle crisi parziali ha suggerito che un POTENZIAMENTO dell’AZIONE del GABA
possa ridurre l’eccitabilità neuronale e aumentare così la soglia epilettogena.
Il
GABA, identificato per la prima volta nel 1950 è il neurotrasmettitore
inibitorio più diffuso del SNC.

Esso
viene formato per decarbossilazione dell’acido glutammico catalizzata dalla GAD
(glutammato decarbossilasi), enzima citosolico altamente specifico che ha come
cofattore il piridossal fosfato (Vit B6), viene accumulato all’interno delle
vescicole da un trasportatore attivo specifico.
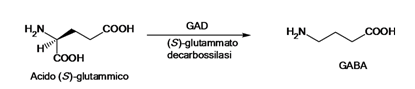
Dopo
il rilascio dalle vescicole mediato dal classico meccanismo Ca+2-dipendente
la sua azione viene bloccata con un
reuptake attivo e una degradazione operata dall’enzima GABA-α-chetoglutaricotransaminasi (GABA T) che
lo deammina a semialdeide succinica la quale viene ossidata ad acido succinico
ad opera di una semialdeide-succinico-deidrogenasi NAD-dipendente e
infine entra a far parte del ciclo di Krebs;
il gruppo amminico, invece, viene trasferito dalla GABA-T ad una
molecola di α-chetoglutarato per formare l'acido
glutammico che viene riutilizzato per la sintesi di nuovo GABA.
Studi
elettrofisiologici e biochimici hanno dimostrato che per esplicare la sua
azione, il GABA deve legarsi a due tipi di recettori GABAA e GABAB
che differiscono fra loro per profilo farmacologico, struttura molecolare
e meccanismo di trasduzione del segnale.
Quando
il GABA si lega a recettori GABAB, essendo questi dei recettori
accoppiati alle proteine G, viene attivata la proteina G inibitrice (Gi) che
produce una inibizione dell’enzima adenilato ciclasi. La conseguente riduzione
della concentrazione di cAMP si traduce in una inibizione dei canali Ca+2
dipendenti implicati nel rilascio dei neurotrasmettitori.
Il
recettore GABAA è un recettori canale permeabile allo ione Cl-
che a livello macromolecolare si presenta come un pentamero costituito da due
subunità α, due subunità β e una subunità γ nel quale sono presenti i siti di legame
specifici per le seguenti molecole:
1) Il sito di legame per il GABA è situato sulla
subunità β e la sua attivazione si traduce nell'apertura del
canale ionico con conseguente iperpolarizzazione della membrana
2) Il sito di legame per le benzodiazepine, situato
sulla subunità α, viene riconosciuto
anche da ligandi ad azione antagonista competitiva (flumazenil). Questo sito,
quando viene attivato ha la capacità di modulare allostericamente,
rispettivamente facilitando e inibendo, l’interazione del GABA con il proprio
sito di legame con conseguente attivazione o riduzione dell’attività del
canale.
3) Il sito di legame per i barbiturici si trova
all’interno del canale per lo ione Cl- che viene in questo modo
attivato: i barbiturici, al contrario delle benzodiazepine, sono perciò capaci
di indurre influsso di cloro indipendentemente dal legame
del GABA con il recettore
Il recettore GABAA è un importante
sito d'azione anche per molti anestetici generali, per l'etanolo e per numerosi
derivati steroidei. In particolare, questi ultimi composti sembrano possedere
dei siti di legame specifici a livello del canale ionico.
Molti
dei farmaci anticonvulsivanti clinicamente efficaci attivano il recettore GABAA,
aumentando il flusso di ioni Cl- nella cellula e iperpolarizzando
così il neurone. Tra questi abbiamo: Fenobarbitale, Primidone, alcune
benzodiazepine, Vigabatrin, Tiagabina e Gabapentina.

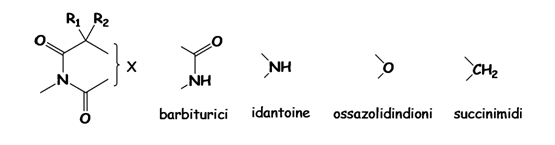
Da
esso si è sviluppata poi una intensa ricerca, tesa soprattutto a cercare di
scindere l’azione anticomiziale da quella sedativa, basti pensare che le
strutture chimiche di gran parte dei farmaci introdotti in commercio prima del
1965 (idantoine, ossazolidindioni e succimidi) sono strettamente correlate alla
struttura del fenobarbitale (DERIVATI UREIDICI)
Esso
mima e aumenta le azioni del GABA sul complesso recettoriale GABAA
portando così ad una iperpolarizzazione della membrana e quindi ad una minor
suscettibilità all’innesco del potenziale d’azione.
Il
fenobarbitale presenta un assorbimento orale completo ma lento e si ritrova nel
sangue legato per il 40-60% alle proteine plasmatiche. Fino al 25% della dose
viene eliminato mediante escrezione renale in forma immodificata (essendo una
acido debole la sua ionizzazione aumenta nelle urine alcaline cosicché si ha un
minor riassorbimento e quindi una maggiore eliminazione), mentre il restante
75% viene inattivato dal CYP2A9. Il farmaco è capace di indurre a
livello epatico le isoforme CYP e l’uridin difosfato glucuronil-transferasi
(UGT) per cui farmaci che vengono metabolizzati con questi enzimi
(contraccettivi orali, steroidi, warfarin, antidepressivi triciclici, altri
antiepilettici come la fenitoina) possono venir degradati più rapidamente
quando co-somministrati al fenobarbitale.
La
sua efficacia nelle forme parziali e tonico-cloniche generalizzate, la scarsa
tossicità e il basso costo ne fanno un farmaco di scelta in questi tipi di
epilessia anche se il suo impiego è stato ridotto a causa degli effetti
sedativi che compaiono in tutti i pazienti all’inizio della terapia ma che
possono andar incontro a tolleranza e dalla
tendenza a dare iperattività nei bambini e confusione negli anziani.
A
dosaggi eccessivi, compaiono nistagmo, atassia, arresto respiratorio
analogamente a tutti gli altri barbiturici, nei quali gli effetti deprimenti a
livello del SNC aumentano progressivamente con l’aumentare della dose.
Un analogo del fenobarbitale in cui l’ossigeno carbonilico è sostituito
con due atomi di idrogeno è il PRIMIDONE (Mysoline®),

la
cui efficacia nelle forme parziali e tonico-cloniche generalizzate è
attribuibile ai suoi metaboliti, fenobarbitale e feniletilmalonammide (PEMA).
Oltre che per meccanismo d’azione, il farmaco somiglia al fenobarbitale anche
negli effetti collaterali che vanno dalla sedazione al nistagmo, atassia e
vertigini.
Anche
le benzodiazepine condividono con il fenobarbitale la capacità di potenziare
l’azione del GABA ma con un meccanismo diverso dovuto al legame in punti
diversi del recettore GABAA: il fenobarbitale, infatti, legandosi
direttamente al recettore ne prolunga il tempo di apertura del canale, mentre
le BDZ legandosi a un sito regolatorio del recettore diverso da quello di
legame per il GABA, aumentano allostericamente il legame del neurotrasmettitore
al canale.
Tutte
le BDZ possiedono proprietà antiepilettiche ma solo CLONAZEPAM (Frisium®)
e CLOBAZAM (Rivotril®)
sono stati approvati per il trattamento a lungo termine perché inducono una
minore sedazione.

Vengono
usati nella terapia delle assenze anche se i loro effetti sviluppano tolleranza
in 1-6 mesi di terapia dopo i quali alcuni pazienti non rispondono più ad essi
indipendentemente dal dosaggio.
La
tolleranza, cioè il graduale aumento della dose necessaria a produrre l’effetto
richiesto, è meno marcata di quella indotta dai barbiturici ed è causata da un
meccanismo diverso: la tolleranza da barbiturici è di natura farmacocinetica
essendo dovuta al loro effetto induttivo sugli enzimi microsomiali epatici
farmacometabolizzanti, mentre quella da BDZ è farmacodinamica in quanto sembra
conseguente ad una down regulation
recettoriale (= diminuzione del numero dei recettori o della risposta del
recettore a seguito della persistenza dell’agonista).
Inoltre,
la brusca interruzione nell’impiego anticomiziale può dare un peggioramento
delle crisi o addirittura uno STATO di MALE EPILETTICO o STATUS EPILEPTICUS,
cioè una crisi epilettica che dura più di 30 minuti oppure due o più crisi
durante un periodo di 30 minuti senza un
completo recupero tra di esse, che può indurre ipossia, ipotensione, acidosi e
ipertermia.
Questo
stato è una grave emergenza medica e la terapia deve risolvere le crisi entro
60 minuti. In genere si impiegano LORAZEPAM (Tavor®) e DIAZEPAM (Noan®;
Valium®)
per via endovenosa in quanto presentano una azione rapida. Il diazepam per via
orale viene anche usato in occasione di attacchi febbrili per prevenire le
convulsioni nei bambini ad alto rischio.
Oltre
che agendo direttamente sui recettori, il potenziamento dell’azione del GABA si può ottenere anche riducendo
la sua degradazione o inibendo il suo reuptake.
La gabapentina, la vigabatrina e la tiagabina sono tutti analoghi strutturali
del GABA.
La
GABAPENTINA
(Neurotin®)
è un farmaco antiepilettico approvato dalla FDA nel 1994 per la terapia delle
crisi parziali anche generalizzate, specie in associazione con altri farmaci.

L’assorbimento
intestinale dipende dal trasporto per gli amminoacidi e quindi mostra la
caratteristica saturabilità: aumentando la dose la quantità assorbita non
aumenta in modo proporzionale e ciò rende il farmaco privo di effetti
collaterali associati al sovradosaggio.
Gli
effetti collaterali più comuni in terapia sono sonnolenza, vertigini, atassia e
affaticabilità che in genere spariscono entro due settimane dall’inizio della
terapia cronica. Inoltre, il farmaco non dando interazioni farmacologiche, è
ottimo per l’impiego in associazione.
Viene
anche usata per l’emicrania, il dolore cronico (adiuvanti) e il disturbo
bipolare.
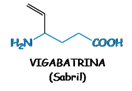
strutturalmente simile al GABA con
un vinile in γ, capace di inibire specificatamente e irreversibilmente mediante
legame covalente, la GABA transaminasi, enzima responsabile della
metabolizzazione del GABA, aumentando così il contenuto di GABA nel cervello.
Nonostante l’emivita plasmatica breve, il
vigabatrin produce un effetto a lunga durata a causa dell’inibizione enzimatica
irreversibile (ci vogliono 3 giorni per la rigenerazione dell’enzima) e ciò
permette una somministrazione per via orale al giorno nel trattamento delle
crisi parziali refrattarie ad altri farmaci e negli spasmi infantili. Gli
effetti collaterali sono in genere di tipo psichiatrico e neurologico; in
particolare provoca retinopatie gravissime il che ne limita l’uso.
Nello
stesso anno del Vigabatrin, è stato approvato l’uso da parte della FDA della TIAGABINA
(Gabitril®),

Studi
elettrofisiologici condotti sui neuroni durante una crisi parziale hanno
rivelato che una caratteristica peculiare di essi è una scarica di pdA ad alta
frequenza generalmente rara durante la fisiologica attività neuronale; da qui
l’idea che usando sostanze in grado di ridurre l’eccitabilità neuronale si
potessero inibire le scariche e ridurre le crisi.
Alcuni
dei più importanti farmaci usati nel prevenire le crisi parziali e
tonico-cloniche generalizzate agiscono attraverso il BLOCCO dei CANALI del Na+.
I
CANALI del Na+ sono proteine integrali di membrana che permettono
l’ingresso di ioni Na+
all’interno delle cellule su cui sono presenti, ossia tutte le cellule
eccitabili. Questi canali sono caratterizzati da cinetiche di attivazione e
deattivazione estremamente rapide (1-10 msec) e sono formati da una subunità
fondamentale, la subunità α,
e da subunità β accessorie.
La
subunità α è la responsabile
di tutte le proprietà elettrofisiologiche e farmacologiche di questi canali.
Essa è il cuore del canale ed è formata da 4 domini uguali, etichettati I, II,
III e IV, che si associano a formare un tetramero. Ciascun dominio è formato da
6 α eliche di transmembrana
(S1-S6).

Tra
queste α eliche di
transmembrana, la regione S4 agisce come sensore di voltaggio del canale e la
sua sensibilità al voltaggio è dovuta alla presenza di residui carichi
positivamente (Arg) ogni tre amminoacidi idrofobici: quando viene stimolata da
un cambiamento nel voltaggio transmembranario, questa regione si muove verso il
lato extracellulare della membrana, rendendo così il canale permeabile agli
ioni. Gli ioni Na+ vengono così trasportati passivamente attraverso
il poro che è la parte responsabile della selettività agli ioni, essendo di
larghezza approssimativa di 0.3-0.5 nm il che consente l’attraversamento di un
solo ione Na+ associato ad una molecola d’acqua, e formato di
amminoacidi carichi negativamente. Altra regione importante in questo tipo di
canale è quella che unisce i domini III e IV, in quanto è la regione che
scollega il canale dopo attivazione prolungata, inattivandolo. Questa regione
funziona, infatti, da cancello in grado di chiudere dal lato intracellulare il
canale, impedendo l’ingresso del Na+ nella cellula.
Nell'assone
a riposo il cancello e' chiuso; durante il potenziale d’azione il cancello si
apre per permettere l’attivazione del canale e la conseguente entrata di un
discreto numero di ioni Na+ a causa del loro gradiente chimico, che
genera così una depolarizzazione e aumento dell’eccitabilità della cellula.
Dopo
la depolarizzazione, il cancello si chiude e lo stato di inattivazione si
conclude quando il potenziale di membrana della cellula si ripolarizza.
Pertanto,
essendo questi canali regolati dalla differenza di potenziale, svolgono un
ruolo fondamentale nella trasmissione del potenziale d’azione: l’attivazione di
questi canali, conseguente all’aumento del potenziale di membrana, porta
all’entrata di un discreto numero di ioni Na+ per il loro gradiente
chimico, causando così una depolarizzazione e aumento dell’eccitabilità della
cellula.

Le
molecole che agiscono da bloccanti del canale del Na+ sono la
Fenitoina, la Carbamazepina, l’Oxcarbamazepina e l’acido valproico, e la loro azione
è voltaggio e frequenza dipendente in quanto essi vanno a bloccare
preferenzialmente l’eccitabilità di cellule già stimolate in maniera ripetitiva
senza interferire con i neuroni attivi a bassa frequenza durante il loro
normale stato di attività e il blocco è tanto maggiore quanto maggiore è la
frequenza di eccitazione. Questa proprietà deriva dalla loro capacità di
legarsi preferenzialmente ai canali del Na+ nel loro stato inattivo,
prevenendo così il ritorno allo stato di riposo con conseguente prolungamento
del periodo refrattario.
La FENITOINA (Aurantin®) è il membro più importante della famiglia delle
IDANTOINE, nate dalla ricerca di analoghi non sedativi del fenobarbitale.
Come
per il fenobarbitale, l’azione anticonvulsiva è legata alla presenza di un
fenile o di un altro sostituto aromatico in 5, mentre dalla SAR si è visto che
la presenza di un gruppo alchilico aumenta le proprietà sedative.
La
fenitoina ha però alcune proprietà farmacocinetiche che devono essere
considerate attentamente nel suo impiego clinico. Mentre a causa della sua
scarsa idrosolubilità viene difficilmente somministrata per via endovenosa (e
ciò ha portato alla produzione della FOSFENITOINA, un profarmaco idrosolubile
che viene convertito in fenitoina ad opera delle fosfatasi epatiche e
eritrocitarie), dopo somministrazione orale è ben assorbita e per il 90% si
ritrova nel sangue legata alle proteina plasmatiche, in primis
all’albumina. Dato che alcuni farmaci come i salicilati e l’acido valproico
inibiscono questo legame in maniera competitiva, essi andranno a provocare in
caso di co-somministrazione un aumento di concentrazione libera, fenomeno che
si verifica anche nei neonati e nei pazienti con ipoalbuminemia, per i quali,
il fegato, sede di sintesi delle proteine plasmatiche, è immaturo o
inefficiente.
L’aumento
di fenitoina libera provoca anche un aumento della sua clearance epatica che
avviene ad opera del CYP2C9 e del CYP2C10. Inoltre, essendo la molecola un induttore
enzimatico, può incrementare il metabolismo di altri farmaci (anticoncezionali,
warfarin) e, allo stesso modo, il suo metabolismo, può venir aumentato o ridotto
competitivamente da altri composti che si avvalgono degli stessi enzimi
epatici: ad esempio, il valproato incrementa i livelli di fenitoina libera
oltre che attraverso la competizione con le proteine plasmatiche anche
attraverso l’inibizione metabolica, mentre il fenobarbitale e l’etanolo posso
provocare un iniziale aumento della attività del farmaco per inibizione
competitiva a livello citocromiale seguita da una riduzione per lo stabilirsi
dell’induzione enzimatica.
Da tutto
ciò (legame con le proteine plasmatiche, non linearità della cinetica di
eliminazione, metabolismo saturabile e inducibile) deriva che gli effetti della
fenitoina possono aumentare o diminuire in maniera imprevedibile e poiché
l’intervallo di concentrazione plasmatica nel quale essa raggiunge l’efficacia
senza causare effetti collaterali è ristretto, è necessario sempre il
monitoraggio delle sue concentrazioni plasmatiche.
Gli
effetti collaterali meno gravi come vertigini, atassia, cefalea e nistagmo
iniziano a concentrazioni plasmatiche maggiori di 100 µmol/L e possono divenir
gravi a concentrazioni maggiori di 150 µmol/L, mentre gli effetti tossici più
gravi sono le aritmie cardiache associate o meno a ipotensione e/o depressione
del SNC, che si verificano più frequentemente nei soggetti anziani e
cardiopatici ma possono svilupparsi anche nei soggetti giovani e sani.
L’iperplasia
gengivale, più deturpante che dannosa, è un effetto collaterale che si sviluppa
gradualmente ed è legata ad una alterazione del metabolismo del collagene, così
come l’irsutismo che probabilmente origina da un aumento della secrezione di
ormoni androgeni.

Essa agisce
andando a bloccare selettivamente la scarica neuronale ad alta frequenza senza
interferire con quella fisiologica attraverso l’inibizione voltaggio e
frequenza dipendente dei canali del Na+.
La
carbamazepina viene usata anche nel trattamento di vari tipi di dolore
neuropatico, compresa la nevralgia del trigemino, una condizione estremamente
dolorosa originata da una scarica parossistica dei neuroni associati alla via
sensoriale del trigemino e pertanto coinvolgente meccanismi neuronali simili a
quelli delle crisi epilettiche.
Dopo
somministrazione orale, questo composto scarsamente idrosolubile, viene ben
assorbito e si distribuisce in tutti i tessuti. Nell’uomo, la carbamazepina
viene principalmente metabolizzata dal CYP3A4 a 10,11-epossido, un metabolita
attivo la cui concentrazione plasmatica può raggiungere anche il 50% di quella
della molecola madre, in particolare se somministrata in associazione con
induttori come fenitoina e fenobarbitale. La stessa carbamazepina è un induttore
enzimatico per cui può aumentare il metabolismo di altri farmaci
co-somministrati.
Essa
provoca una varietà di effetti collaterali che includono sonnolenza (minore di
quella indotta da fenitoina), vertigini, atassia che sono di minor entità se
sono impiegate preparazioni a lento rilascio che evitano il raggiungimento di
elevati livelli plasmatici dopo una singola dose.
All’inizio
della terapia si sviluppa in circa il 10% dei pazienti una leucopenia lieve e
transitoria che generalmente si risolve entro i primi 4 mesi di terapia
continuativa, mentre nel 2% dei casi può diventare persistente e richiedere la
sospensione del farmaco.
Un chetoanalogo della carbamazepina, l’OXCARBAMAZEPINA
(Tolep®) viene usato come farmaco aggiuntivo nel trattamento delle forme
parziali di epilessia.


Dopo
somministrazione orale, viene assorbito in modo rapido e completo, andandosi a
legare per il 90% alle proteine plasmatiche.
I suoi
principali enzimi metabolizzanti sono il CYP2C9 e CYP2C19 verso i quali ha
un’azione inibitoria. Pertanto, il valproato può ritardare il
metabolismo di altri farmaci co-somministrati e substrati di questi enzimi,
aumentando la loro concentrazione plasmatica così come legandosi all’albumina
può spiazzare altri farmaci dal legame con questa proteina plasmatica.
In circa
il 40% dei pazienti sotto terapia con valproato si sono riscontrati aumenti
degli enzimi epatici che richiedono il monitoraggio della funzionalità di
questo organo. Si sono riscontrati anche disturbi gastrointestinali, aumento di
peso, sedazione, atassia e tremore che normalmente rispondono ad una riduzione
della dose.
Come
fenitoina e carbamazepina, il valproato allunga il tempo di ripresa dei canali
del Na dall’inattivazione, tuttavia la sua azione antiepilettica sembra esser
legata anche ad un potenziamento dell’azione del GABA sia attraverso una
stimolazione dell’ enzima di sintesi sia attraverso l’inibizione degli enzimi
degradativi di questo neutrotrasmettitore.
Inoltre,
il valproato riduce lievemente la corrente del Ca+2 e ciò spiega la
sua efficacia ne controllare sia le crisi parziali e tonico-cloniche che le
assenze.
Infatti la
caratteristica elettroencefalografica delle assenze consiste nella presenza di
scariche generalizzate PUNTA-ONDA a una frequenza di 3 Hz, localizzate nel
talamo e nella neocorteccia: questi ritmi a bassa frequenza sono resi possibili dalla presenza nei
neuroni talamici di un particolare tipo di corrente voltaggio dipendente del Ca+2,
la corrente a bassa soglia T.
I CANALI
del CALCIO sono proteine integrali di membrana che formano canali ionici in
grado di condurre i cationi calcio attraverso la membrana plasmatica; la loro
apertura può avvenire a causa di un cambiamento di voltaggio (canali del calcio
voltaggio dipendenti) oppure a causa del legame di una sostanza (canali calcio
ligando dipendenti)
La
struttura molecolare dei canali calcio voltaggio dipendenti è del tutto
simile a quella dei canali del Na
voltaggio dipendenti, sebbene piccole differenze nelle sequenze amminoacidiche cambino
profondamente la selettività. La classificazione dei canali voltaggio
dipendenti per il Ca è effettuata in base alle proprietà elettrofisiologiche
in: i canali a bassa soglia (transient)
o canali T che si attivano in seguito a piccole depolarizzazioni e si
inattivano molto rapidamente, mediando l’ingresso di calcio nei neuroni e
quindi controllando varie funzioni calcio-dipendenti; i canali ad alta soglia
(long lasting) si attivano in seguito a forti depolarizzazioni e rimangono
aperti più a lungo e sono suddivisi a loro volta in vari sottotipi, dei quali
il sottotipo L è importante nella regolazione della contrazione della
muscolatura liscia e del miocardio e i sottotipi N e P sono importanti nel rilascio
di neurotrasmettitori ed ormoni.
Nei
neuroni talamici è la corrente T che media le scariche punta-onda tipiche delle
assenze, per cui il meccanismo d’azione dei farmaci usati in questa forma di
epilessia, come il valproato e l’etosuccimide, consiste proprio nel BLOCCO DEI CANALI DEL CA+2 che
mediano tali correnti.
L’ETOSUCCIMIDE
(Zarotin®) appartiene alla classe delle SUCCIMIDI ed è un altro di quei farmaci
sviluppati empiricamente modificando la struttura dell’anello dell’acido
barbiturico.

I più
comuni effetti collaterali dose-dipendente sono disturbi gastrointestinali
(nausea, vomito, anoressia), sonnolenze o euforia, tutti effetti ai quali si
sviluppa con il tempo una certa tolleranza.
Negli
ultimi anni la ricerca farmaceutica sui composti impiegati nell’epilessia ha
ripreso piede con l’introduzione in terapia del:
Il TOPIRAMATO
(Topamax®) ha un’ampia attività antiepilettica andando a bloccare i canali del
Na e viene impiegato anche per il trattamento delle cefalee.

Il LEVETIRACETAM
(Keppra®) è un analogo strutturale del piracetam il cui meccanismo d’azione non
è ancora del tutto chiaro. Si pensa che
possa andare ad agire con i canali del Ca+2

Per quanto riguarda la terapia non farmacologica, la
stimolazione del nervo vago è un approccio terapeutico recente che è indicato
in casi di epilessia farmacoresistente in cui la terapia chirurgica sia non
possibile o controindicata. La sua efficacia è inferiore a quella della terapia
chirurgica ma è stata dimostrata in una serie di studi clinici. Richiede
l'impianto di un elettrostimolatore che viene collegato con il nervo vago
sinistro il quale trasporta le afferenze sensorie dai visceri al cervello. Per
un meccanismo ancora sconosciuto la modulazione terapeutica della sua attività
elettrica influenza l'attività elettrica cerebrale in modo da rendere il
cervello meno suscettibile alla formazione di focolai epilettici. Come la
terapia chirurgica, la stimolazione del nervo vagale richiede l'assistenza da
parte di centri specializzati.
Le
principali attività della ricerca sull'epilessia sono concentrate sulla
scoperta di farmaci ancora più efficaci e sempre meglio tollerati anche con il
mezzo di modelli sperimentali sempre più raffinati. Inoltre, verranno provati
nuovi protocolli di elettrostimolazione e l'applicazione di farmaci antiepilettici
tramite sonde intracerebrali che rilasciano il farmaco solo nella regione in
cui originano le crisi epilettiche. In tal modo si potrebbe raggiungere un
effetto specifico evitando gli effetti collaterali del farmaco sul tessuto
cerebrale sano.
Fu durante la mia ricerca sull'HIV / Herpes che mi sono imbattuto nelle informazioni Hiv / Herpes; informazioni che è abbastanza facile da trovare quando si effettua una ricerca di STD su google. Ero in cospirazione, al momento pensavo che l'HIV / Herpes Cured "essere una cospirazione fosse qualcosa di ignorante, ma ho trovato abbastanza interessante sulla medicina erboristica. Ho fatto delle domande sulla cura delle erbe sui siti ufficiali di HIV / Herpes e sono stato messo al bando dai moderatori che mi hanno detto che stavo pappagliando la propaganda Hiv / Herpes. Questo ha rafforzato la mia convinzione che esiste una cura per Hiv / Herpes Poi ho trovato una donna dal nome della Germania Achima Abelard Dr Itua Cura il suo Hiv quindi gli mando una mail sulla mia situazione, poi ne parlo di più e mandami la sua medicina erboristica che ho bevuto per due settimane. E oggi sono Cured no Hiv / Herpes nella mia vita, ho cercato gruppi di Hiv / Herpes per tentare di entrare in contatto con le persone per saperne di più su Hiv / Herpes Herbal Cure's ho creduto in questo momento che tu con la stessa malattia questa informazione è utile per te e volevo fare il meglio che potevo per diffondere queste informazioni nella speranza di aiutare altre persone. Che Dr Itua Herbal Medicine mi fa credere che ci sia una speranza per le persone che soffrono, la malattia di Parkinson , Schizofrenia, Cancro, Scoliosi, Fibromialgia, Sindrome da tossicità da fluorochinolone Fibrodisplasia Ossificante Progressiva.Infertilità, Epilessia, Diabete, Celiachia, Artrite, Sclerosi laterale amiotrofica, Morbo di Alzheimer, Carcinoma adrenocorticale.Astma, Malattia allergica s.Hiv_ Aids, Herpes, malattia infiammatoria intestinale, Copd, diabete, epatite, ho letto su di lui online come cura Tasha e Tara, Conley, Mckinney e molti altri suffrin da ogni tipo di malattia, così l'ho contattato. È un medico erborista con un cuore unico di Dio, contatta Emal..drituaherbalcenter@gmail.com Phone o whatsapp .. + 2348149277967.
RispondiElimina